Follow Us
Cystic fibrosis is a life-altering genetic disorder that impacts various organs, particularly the lungs and digestive system. This inherited condition affects millions worldwide, causing thick, sticky mucus to accumulate in the body. Understanding cystic fibrosis is crucial for those affected and their families to control cystic fibrosis symptoms & improve their quality of life.
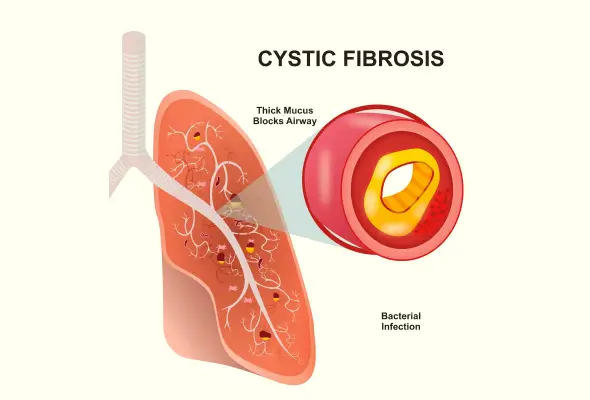
Cystic fibrosis is an inherited disorder running in families. It affects various body organs, particularly the lungs, pancreas, and digestive system. This inherited condition occurs when a person receives a faulty gene from both parents, leading to the generation of thick, sticky mucus that clogs tubes, ducts, and airways.
Cystic fibrosis has an impact on various organs, causing a range of symptoms that vary from person to person. These may include:
Other Cystic Fibrosis Disease Symptoms:
Cystic fibrosis is a common genetic disease occurring as a consequence of alterations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene.
This gene impacts the production and function of the CFTR protein. This protein regulates water and salt movement in and out of cells. When the CFTR protein is faulty or absent, it causes the secretion of thick, sticky mucus that clogs various organs.

To develop cystic fibrosis, a child must inherit two copies of this mutated CFTR gene, one from each parent. If an individual inherits only one copy, they become a carrier but do not typically show disease symptoms. Carrier individuals can pass the mutated gene to their children.
The risk of having a child with cystic fibrosis increases when both parents are carriers of the mutated gene. In such instances, there is a 25% chance that their child will develop cystic fibrosis, a 50% chance the child will be a disease carrier and a 25% chance the child will neither be a disease carrier nor have the disease.
Some ethnic groups show higher instances of carrying the cystic fibrosis gene. Individuals of Northern European descent, especially those of Ashkenazi Jewish heritage, are more likely to be carriers. However, cystic fibrosis can affect individuals from all racial and ethnic backgrounds.
Cystic fibrosis has an impact on various organ systems, resulting in a range of complications that can worsen over time.
Diagnosing cystic fibrosis involves several steps and tests to confirm this genetic disorder. These may include:
While there is no cure, advancements in medical care have significantly enhanced the outlook for individuals with this condition.
It is crucial to seek medical attention if new or worsening symptoms appear. These may include:
Cystic fibrosis cannot be prevented, as it is an inherited genetic disorder. However, there are steps that individuals can take to reduce the risk of having a child with cystic fibrosis.
Cystic fibrosis can significantly impact the lives of those affected, challenging both patients and doctors. The complexity of this genetic disorder requires a comprehensive approach to manage symptoms & improve quality of life. Advances in diagnosis, treatment, and care have led to better outcomes and longer life expectancy for individuals with cystic fibrosis. These improvements offer hope and support to patients and their families as they navigate the challenges of living with this condition.
While a cure for cystic fibrosis remains elusive, ongoing research and medical breakthroughs continue to enhance our understanding and management of the disease. The development of targeted therapies, such as CFTR modulators, has opened new avenues to treat the underlying cause of cystic fibrosis. As we look to the future, the focus remains on further improving cystic fibrosis treatments, supporting patients, and ultimately finding a cure for this complex genetic disorder.
Cystic fibrosis is due to a faulty gene inherited from both parents. This defective gene changes how salt moves in and out of cells, resulting in thick, sticky mucus in the respiratory, digestive, and reproductive systems. For a child to have cystic fibrosis, they must inherit one copy of the mutated CFTR gene from each parent.
Currently, there is no cure available for cystic fibrosis. However, advancements in treatment have improved the quality of life & life expectancy in individuals with this condition. Various therapies and medications are available to manage symptoms, prevent complications & make the condition easier to live with.
The child having both parents who carry the abnormal CF gene is at risk.
Cystic fibrosis affects both males and females. However, some studies suggest that females with cystic fibrosis may experience more severe symptoms and complications compared to males. Women tend to have more lung infections and often develop symptoms earlier in life. Despite this, the condition itself is not more common in one gender than the other.